News & Events
News & Events
News
May 25, 2018
Destabilized cells cause pancreatic cancer in mice
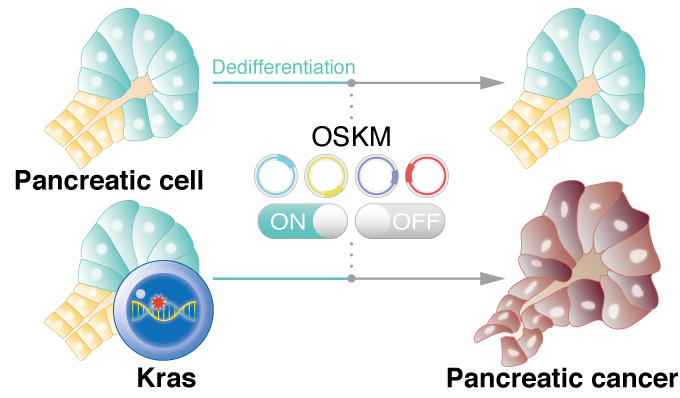
Reprogramming pancreatic cells with a mutation in Kras gene promotes cancer in mice, suggesting dedifferentiation is a trigger for the cancer onset.
CiRA researchers show that briefly initiated reprogramming in cells with a Kras gene mutation triggers the development of pancreatic cancer. The study, which can be read in Nature Communications, suggests that dedifferentiation is a vital first step to stimulating cells with the Kras mutation into cancer. The findings provide a new theory for why pancreatic cancer is one of the deadliest and most difficult to treat.
Among cancers, pancreatic cancer has the worst outcome, with only 20% of patients expected to survive one year after the diagnosis. Pancreatic cancer is also unusual in that mutations in the Kras gene can be found in more than 90% of cases, while no other mutation comes close to this frequency in other cancers.
Yet mutations are not enough for the cancer development. University of Tokyo Professor Yasuhiro Yamada, who led the study while at CiRA, says that in many animal models, there is "a substantial latency period for cancer development, indicating that additional events are required."
Yamada has been using iPS cell reprogramming technology to identify these additional events.
No genetic mutations are required for a cell to be reprogrammed to an iPS cell, but major epigenetic changes must occur. Epigenetics describe modifications to the gene, like the attachment of molecules, which expose or conceal the gene to external signals. A crucial first step in reprogramming controlled by epigenetics is dedifferentiation, or the loss of cell identity. Interestingly, dedifferentiation is also frequently seen in cancers.
To study pancreatic cancer, Yamada and his colleagues developed mutant mice in which they reprogram pancreatic cells that express the Kras mutation. The pancreas is made up of acinar and ductal cells. They found that by activating the reprogramming process transiently in acinar cells with the Kras mutation, so as not to completely reprogram the cells to iPS cells, resulted in pancreatic ductal adenocarcinoma, a type of pancreatic cancer.
Looking for a molecular explanation, CiRA Associate Professor Takuya Yamamoto, who contributed to the study, found that the transient reprogramming lowered the acetylation of H3K27 on crucial genes.
"The H3K27cetylation is used as a marker of enhancers," he said. Acinar cell enhancers are genes responsible for stabilizing acinar cell identity.
The lost activity of the enhancers caused the acinar cells to dedifferentiate. Conversely, the cancer could be avoided by forcing the activity of the enhancers despite the transient reprogramming process.
Even if the dedifferentiation is only brief, it is enough time for the Kras mutation to activate ERK, a molecule associated with cell proliferation. Consequently, ERK remained activated even when the reprogramming was turned off, which would explain the adenocarcinoma.
Finally, treating the mice with caerulein, a known inflammatory stimulant of the pancreas, also repressed the acinar cell enhancers and led to cancer development. Yamada wonders whether this finding could explain why pancreatic cancer returns in a large proportion of patients following surgery.
"We connected the repression of enhancers with inflammation," he said. "This is totally speculation, but the operation causes inflammation. That may explain a part of recurrence of pancreatic cancer."
Paper Details
- Journal: Nature Communications
- Title: In vivo reprogramming drives Kras-induced cancer development
- Authors: Hirofumi Shibata1,2, Shingo Komura1, Yosuke Yamada1,3, Nao Sankoda1, Akito Tanaka1, Tomoyo Ukai1, Mio Kabata1, Satoko Sakurai1, Bunya Kuze2, Knut Woltjen1,4, Hironori Haga3, Yatsuji Ito2, Yoshiya Kawaguchi1, Takuya Yamamoto1,5, and Yasuhiro Yamada1,5,6
- Author Affiliations:
- Center for iPS Cell Research and Application (CiRA), Kyoto University, Kyoto 606-8501, Japan
- Department of Otolaryngology, Head and Neck Surgery, Gifu University Graduate School of Medicine, Gifu, 501-1194, Japan
- Department of Diagnostic Pathology, Kyoto University Hospital, Kyoto 606-8501, Japan
- Hakubi Center for Advanced Research, Kyoto University, Kyoto 606-8501, Japan
- AMED-CREST, AMED, Tokyo, 100-0004, Japan
- Center for Experimental Medicine and Systems Biology, Institute of Medical Science, University of Tokyo, Tokyo, 108-8639, Japan