Research Activities
Research Activities
Publications
April 02, 2015
An iPS cell model to study muscle degeneration in infants
Spinal muscular atrophy (SMA) is an autosomal disease that is often recognized in the first six months of life and is considered the leading genetic cause of infant mortality. Patients suffer from progressive muscle weakness and eventual death. While some patients can survive for decades with the assistance of an artificial respirator, quality of life is severely compromised. It is a ravaging disease with no treatment. "I have managed a couple of SMA patients. They are intellectually normal but cannot move," explains Associate Professor Megumu Saito, who led a project that included five different CiRA labs and has resulted in a recent report that gives insight about the early phases of SMA progression using an iPS cell-based model.
Although historically the disease has been diagnosed by a loss of motor neurons, this model suggests that the first defect is due to an absence of acetylcholine receptors (AchR) clustering at the myotubes. The lack of clustering disrupts communication between the neuron and muscle at the neuromuscular junction (NMJ), resulting in atrophy of the muscle and consequent motor neuron death.
At the genetic level, SMA is recognized by insufficient production of the SMN protein. In humans, SMN is synthesized by two gene isoforms SMN1 and SMN2, but with the former producing approximately 90% of the body's SMN levels. However, SMA patients have a deleted SMN1 isoform, resulting in deficient SMN levels. Lead author and neuro-pediatrician Michiko Yoshida therefore investigated ways that could amplify the SMN production from the SMN2 isoform in iPS cells derived from SMA patients. She found that AChR clustering could be improved by treating the cells with valproic acid (VPA), which increases the SMN2 expression, or by applying to them phosphoro-diamidate morpholino oligonucleotides (PMO). These observations, she suggests, "...show SMA is not a neuro-degenerative disease but maybe a developmental one." The implications of this finding argue for genetic tests at the fetal stage, since disease onset will have already progressed by birth.
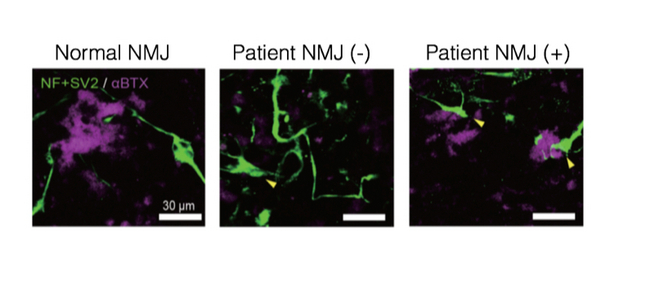
AChR clustering (purple) is clearly present in normal NMJ but absent in SMA patients (-). Treatment with either VPA or PMO (+) recovers the clustering.
Paper Details
- Journal: Stem Cell Reports
- Title: Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs
- Authors: Michiko Yoshida, Shiho Kitaoka, Naohiro Egawa, Mayu Yamane, Ryunosuke Ikeda, Kayoko Tsukita, Naoki Amano, Akira Watanabe, Masafumi Morimoto, Jun Takahashi, Hajime Hosoi, Tatsutoshi Nakahata, Haruhisa Inoue, Megumu K Saito